
Page 36 - 《中国药房》2026年6期
P. 36
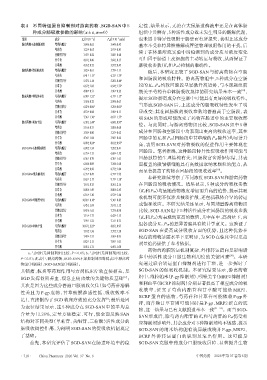
表4 不同转运蛋白抑制剂对游离药物、SGD-SAN 中 5 定性,结果显示,无论在大鼠肠道酶液中还是在离体肠
种成分肠吸收参数的影响(x±s,n=6) 组织中共孵育,5种活性成分既未发生明显的酶解代谢,
组别 成分 K a/(×10 s) P eff/(×10 cm/s) 也未因非特异性吸附于肠壁而有所损失,表明上述成分
-4
-5
游离药物不加抑制剂组 芍药内酯苷 3.84±0.62 3.44±0.42 基本不受非特异性酶解或管壁物理吸附作用的干扰,后
芍药苷 3.22±0.65 2.91±0.81 续于在体肠灌流实验中所检测到的成分质量浓度变化
芹糖甘草苷 3.47±0.82 3.08±0.60
甘草苷 4.10±0.46 3.68±0.17 可归因于肠道上皮细胞的主动转运与吸收,从而保证了
甘草酸 4.26±0.72 3.92±0.47 肠吸收参数(Ka和Peff )检测的准确性。
游离药物+维拉帕米组 芍药内酯苷 3.82±0.61 3.73±1.11 随后,本研究比较了 SGD-SAN 与游离药物在空肠
芍药苷 6.41±1.13 a 6.32±1.39 a 和回肠段的吸收特性。游离药物组中,5 种成分在空肠
芹糖甘草苷 5.47±1.14 5.20±0.44 a
甘草苷 6.67±1.83 6.54±1.72 a 段的 Ka、Peff均较回肠段呈轻微升高趋势,与本课题组前
[18]
甘草酸 4.08±0.51 3.96±0.22 期关于芍药苷单体肠吸收规律的研究结果基本一致 ,
游离药物+吲哚美辛组 芍药内酯苷 6.90±1.21 a 6.17±1.17 a 提示SGD游离成分在空肠中可能具有更好的吸收特性。
芍药苷 3.50±0.72 2.99±0.67
芹糖甘草苷 6.02±0.06 a 5.28±0.05 a 当形成 SGD-SAN 后,上述成分的肠吸收特性发生了明
甘草苷 4.53±0.08 3.90±0.11 显改变:其在回肠段的吸收参数均普遍高于空肠段,表
甘草酸 7.36±1.36 a 6.67±1.37 a 明 SAN 的形成可能改变了药物在肠道中的主要吸收部
游离药物+利血平组 芍药内酯苷 6.38±1.44 a 6.04±0.97 a 位。与此同时,与游离药物组比较,SGD-SAN 组中 5 种
芍药苷 3.16±0.71 2.88±0.60
芹糖甘草苷 3.54±0.80 3.25±0.62 成分在回肠和空肠段中均表现出更高的吸收速率,其在
甘草苷 4.34±1.04 3.98±0.62 回肠中的Ka和Peff (回肠段中甘草酸的Peff除外)均显著升
甘草酸 8.09±0.24 a 8.01±0.97 b 高,表明 SGD-SAN 对药物吸收的促进作用主要体现在
SGD-SAN不加抑制剂组 芍药内酯苷 6.98±1.24 7.28±0.41 回肠段。笔者推测,这种肠段特异性促吸收作用可能与
芍药苷 6.35±1.13 6.68±1.52
芹糖甘草苷 6.36±0.79 6.76±1.63 回肠独特的生理结构有关:回肠段富含派伊尔结,其表
甘草苷 6.84±0.09 7.34±1.60 面覆盖的微皱褶细胞具有高效摄取纳米颗粒的能力,从
甘草酸 6.29±0.50 6.77±1.84 而显著提高了药物在回肠段的吸收效率 。
[19]
SGD-SAN+维拉帕米组 芍药内酯苷 6.35±0.47 6.77±1.92
芍药苷 8.62±2.17 9.17±1.30 a 本研究继续考察了不同浓度SGD-SAN和游离药物
芹糖甘草苷 7.63±0.75 8.36±2.16 在回肠段的吸收情况。结果显示,5 种成分的吸收参数
甘草苷 8.08±1.49 8.80±2.43 (Ka和Peff )均呈随药物浓度增加而升高的趋势,提示其吸
甘草酸 6.16±1.30 6.53±1.46 收机制可能不仅涉及被动扩散,还包括载体介导的转运
SGD-SAN+吲哚美辛组 芍药内酯苷 10.02±0.18 a 9.30±0.85
芍药苷 6.45±1.69 5.67±1.12 或饱和效应。本研究结果还显示,与同剂量游离药物组
芹糖甘草苷 8.42±1.62 7.72±0.90 比较,SGD-SAN组中5种活性成分在回肠段的吸收参数
甘草苷 7.15±1.77 6.42±1.13 (Ka和Peff )均表现出更高的数值,其中在中、高剂量下,两
甘草酸 7.99±1.21 7.31±0.73
SGD-SAN+利血平组 芍药内酯苷 10.67±2.23 a 8.85±0.97 组各成分Ka、Peff的差异普遍具有统计学意义。这验证了
芍药苷 7.10±1.36 5.72±1.20 SGD-SAN 在提高成分吸收方面的优势,且这种优势在
芹糖甘草苷 7.49±1.57 5.96±0.95 较高的药物暴露水平下更明显,为SGD在临床中以更高
甘草苷 8.82±2.13 7.09±1.03 剂量给药提供了参考依据。
甘草酸 8.05±1.71 6.49±1.00
药物的跨膜转运机制复杂,外排转运蛋白是影响诸
a:与同组无抑制剂组比较:P<0.05;b:与同组无抑制剂组比较: [20]
P<0.01;此表中,游离药物、SGD-SAN不加抑制剂组等同表2中游离药 多中药活性成分口服生物利用度的关键因素 。本研
物组(回肠段)、SGD-SAN组(回肠段)。 究通过联合转运蛋白抑制剂进行干预,进一步探讨了
其镇痛、抗炎等药理作用与方剂临床疗效直接相关,是 SGD-SAN 的促吸收机制。本研究结果显示,游离药物
[17]
SGD 发挥益阴养血、缓急止痛功效的关键物质基础 ; 组中,维拉帕米(P-gp抑制剂)、吲哚美辛(MRP2抑制剂)
和利血平(BCRP抑制剂)分别显著提高了相应成分的吸
其次是因为这些成分普遍口服吸收欠佳(如芍药苷溶解
收效率,证实了芍药内酯苷和甘草酸可能是 MRP2、
性差且为 P-gp 底物,甘草酸膜渗透性低、吸收效率不
BCRP 蛋白的底物,芍药苷和甘草苷可能依赖 P-gp 外
足),直接制约了SGD临床疗效的充分发挥 ;最后是因
[3]
排,而芹糖甘草苷则可能同时是 P-gp、MRP2 蛋白的底
为表征结果显示,这 5 种成分在 SGD-SAN 中的平均总 [21―24]
物,这一结果与已有文献报道基本一致 。而当SGD-
含量为 12.26%,定量方法稳定、可行,能全面反映 SAN
SAN 形成后,除芍药内酯苷的 Ka和芍药苷的 Peff仍受相
结构对不同类型(单萜苷、黄酮苷、三萜酸)活性成分的 应抑制剂影响外,其余成分对3种抑制剂均不敏感,提示
肠吸收调控作用,为阐明 SGD-SAN 的促吸收机制奠定 SGD-SAN 的纳米结构能有效屏蔽或绕开 P-gp、MRP2、
了基础。 BCRP 外排转运蛋白的识别及泵出作用。这可能是
首先,本研究评估了 SGD-SAN 在肠道环境中的稳 SGD-SAN 克服中药成分口服吸收屏障、显著提升生物
· 718 · China Pharmacy 2026 Vol. 37 No. 6 中国药房 2026年第37卷第6期