
Page 38 - 《中国药房》2024年24期
P. 38
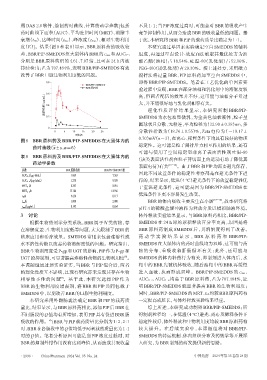
用 DAS 2.0 软件,绘制药时曲线,计算药动学参数[包括 不及1∶1;当PIP浓度过高时,可能会对BBR的吸收产生
药时曲线下面积(AUC)、平均驻留时间(MRT)、消除半 竞争抑制作用,从而会造成BBR的吸收量低的问题。基
衰期(t1/2 )、达峰时间(tmax )、峰浓度(cmax )、相对生物利用 于此,本研究将BBR和PIP的载药质量比确定为1∶1。
度(Fr )]。结果(图 8 和表 8)显示,BBR 原料药的吸收较 本研究通过单因素实验确定空白 SMEDDS 的辅料
差,BBR/PIP-SMEDDS组大鼠体内BBR的cmax和AUC0-t 组成,再通过星点设计-效应面法确定其最优处方为油
分别是 BBR 原料药组的 4.61、7.07 倍,且可在 24 h 内被 酸乙酯(油相)占 18.54%、吐温-80(乳化剂)占 52.16%、
持续检出;Fr为 707.484%,表明 BBR/PIP-SMEDDS 有效 PEG-400(助乳化剂)占29.30%。按上述处方,采用磁力
改善了BBR口服生物利用度低的问题。 搅拌法将过量 BBR、PIP 原料药加至空白 SMEDDS 中,
0.8 即得 BBR/PIP-SMEDDS。笔者在工艺优化的单因素实
BBR/PIP-SMEDDS
BBR原料药
验过程中发现,BBR在部分油相和乳化剂中的溶解度较
0.6
( μg/mL ) 高,但两者配伍的效果并不好,这可能与油相分子量过
大,并不能很好地与乳化剂相容有关。
血药浓度/ 0.4 理化性质评价结果显示,本研究所制 BBR/PIP-
0.2 SMEDDS 为水包油型微乳,为金黄色油状液体,粒子呈
圆球状且分散、无粘连,平均粒径为(32.90±0.38)nm,多
0
0 5 10 15 20 25 分散性指数为(18.76±0.55)%,Zeta 电位为(-19.17±
时间/h
图8 BBR 原料药及 BBR/PIP-SMEDDS 在大鼠体内的 0.70)mV(n=3),在离心、稀释条件下均具有较好的物理
稳定性。这可能是粒子间排斥力相互作用的结果,还有
药时曲线(x±s,n=6)
可能与使用了空间稳定型非离子表面活性剂吐温-80
表8 BBR 原料药及 BBR/PIP-SMEDDS 在大鼠体内的 (该类表面活性剂在粒子剪切面上的运动有助于降低其
药动学参数
表面电荷)有关 [15―16] 。由于BBR和PIP均需要避光保存,
参数 BBR原料药组 BBR/PIP-SMEDDS组
AUC 0-t/(μg·h/mL) 1.069 7.563 因此不同放置条件的稳定性考察都是在避光条件下进
AUC 0-∞/(μg·h/mL) 1.278 9.389 行的,结果显示,低温(4 ℃)避光条件下的放置稳定性优
MRT 0-t/h 8.347 8.916 于室温避光条件,这可能是因为 BBR/PIP-SMEDDS 在
MRT 0-∞/h 13.186 14.956
t 1/2/h 9.634 11.117 低温条件下更不容易发生改变。
t max/h 1.000 2.000 BBR的体内吸收主要发生在小肠 [17―18] ,故本研究将
c max/(μg/mL) 0.142 0.654
pH7.4 的磷酸盐缓冲液作为释放介质以模拟肠液环境。
3 讨论 体外释放实验结果显示,与 BBR 原料药相比,BBR/PIP-
根据生物药剂学分类系统,BBR 属于Ⅳ类药物,存 SMEDDS 在 24 h 时的累积释放百分率更高,原因是将
在溶解度差、生物利用度低等问题,大大限制了 BBR 的 BBR 原料药制成 SMEDDS 后,其溶解度得到了改善。
临床应用和治疗效果。SMEDDS 常用来包裹难溶性或 药 动 学 实 验 结 果 显 示 ,BBR 原 料 药 和 BBR/PIP-
水不溶性药物以改善药物溶解度低的问题。研究指出, SMEDDS在大鼠体内的药时曲线均为双峰,这可能与药
BBR生物利用度受P-gp和UGT的影响,PIP作为P-gp和 物的分布、重吸收和肝肠循环有关;此外,还可能与
[8]
UGT的抑制剂,可显著提高难溶性药物的生物利用度 。 SMEDDS 的体内释药行为有关,即制剂进入体内后,水
本课题组通过预实验证实,当 BBR 与 PIP 混合后,两者 相中的 BBR 先被机体吸收,随后油相中的 BBR 再缓慢
的理化性质互不影响,且现有研究亦未发现其存在生物 进入血液,从而形成双峰。BBR/PIP-SMEDDS 的 cmax、
[8]
相容性不佳的问题 。基于此,本研究选择 PIP 作为 AUC0-t、AUC0-∞均高于 BBR 原料药,Fr为 707.484%,证
BBR 的生物利用度增强剂,将 BBR 和 PIP 共同包载于 明 BBR/PIP-SMEDDS 能显著提高 BBR 的生物利用度;
SMEDDS中,以期改善BBR的口服生物利用度。 同时,BBR/PIP-SMEDDS的MRT、t1/2均较BBR原料药有
本研究采用外翻肠囊法确定 BBR 和 PIP 的载药质 一定提高或延长,与体外释放实验结果呼应。
量比,结果显示,与BBR原料药相比,添加PIP后BBR在 综上所述,本研究成功制得 BBR/PIP-SMEDDS;所
不同肠段的Q值均有所增加,表明PIP具有促进BBR肠 得制剂粒径均一,在低温(4 ℃)避光、离心及稀释条件下
吸收的作用。当BBR与PIP载药质量比分别为1∶2、2∶1 稳定性较好,体外释放和生物利用度均较BBR原料药有
时,BBR在各肠段中的Q值均低于两者载药质量比为1∶1 较大提升。在后续实验中,本课题组将对 BBR/PIP-
时的 Q 值。笔者分析原因可能是当 PIP 浓度过低时,对 SMEDDS的转运机制、体内组织分布及药效学等开展深
BBR的抑制外排作用没有达到峰值,从而造成其吸收量 入研究,为BBR制剂的研发提供新的思路。
· 2996 · China Pharmacy 2024 Vol. 35 No. 24 中国药房 2024年第35卷第24期