
Page 68 - 《中国药房》2022年14期
P. 68
定性离子对分别为 m/z 356.1→356.1、m/z 338.1→338.1; 2.7 CATM中4种立体异构体相对纯度的测定
去簇电压分别为 70、175 V;扫描时间均为 100 ms;碰撞 取 500 ng/mL 目标产物,按“2.3”项下方法进样分
能量均为5 eV。 析,按归一化法计算其相对纯度。
表1 CATM和(S)-2-氧氯吡格雷定性定量分析的梯度 2.8 顺式CATM保留时间的确定
洗脱程序 受试者口服氯吡格雷后1 h,采集前臂静脉血2 mL,
时间/min 流动相A/% 流动相B/% 时间/min 流动相A/% 流动相B/% 置于肝素抗凝管中,即刻以 4 000×g 离心 10 min,取上
0 83 17 23 70 30 层血浆 50 μL,加入乙腈 250 μL,涡旋振荡 5 min,再以
8 83 17 28 0 100
9 80 20 30 0 100 12 000×g离心5 min,取上清液吹干后,以“2.3”项下初始
18 80 20 32 83 17 比例的流动相复溶,取 10 μL,按“2.3”项下方法进样
19 70 30 34 83 17
分析。
2.4 CATM分离纯化的色谱条件
3 结果
以ChromCore 120 C18制备柱(10 mm×50 mm,5 μm)
3.1 CATM的制备情况
为色谱柱,以水为流动相A、乙腈为流动相B进行梯度洗
80 μmol/L的(S)-2-氧氯吡格雷经过2次离体大鼠肝
脱(洗脱程序见表 2);流速为 1 mL/min;进样量为 100
脏灌流生物转化,得灌流液 580 mL,经分离纯化得目标
mL。
产物 2 mg,即 48 μmol 的(S)-2-氧氯吡格雷生物转化得
表2 CATM分离纯化的梯度洗脱程序
5.62 μmol 的目标产物,转化率为 11.71%。分离纯化后
时间/min 流动相A/% 流动相B/% 时间/min 流动相A/% 流动相B/%
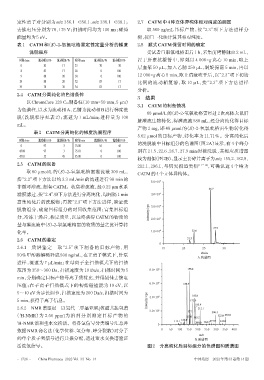
0 97 3 75.00 55 45 的洗脱液中目标组分的色谱图(图2A)显示,有4个峰分
45.00 97 3 75.01 0 100 别在 21.5、22.6、26.7、27.9 min 时被洗脱,其相应质谱图
45.01 55 45 85.00 0 100
较为相似(图2B),显示主要碎片离子为m/z 155.2、182.8、
2.5 CATM的制备 212.1、296.1,与研究报道类似 [11-12] ,可确认这 4 个峰为
取 80 μmol/L 的(S)-2-氧氯吡格雷灌流液 300 mL, CATM的4个立体异构体。
按“2.2”项下方法以约 3.3 mL/min 的流速进行 90 min 的
5.0×10 5
非循环灌流,制备CATM。收集灌流液,经0.22 μm水系
滤膜滤过,按“2.4”项下方法进行分离纯化,每间隔1 min 4.0×10 5
富集纯化后的洗脱物;再按“2.3”项下方法进样,验证洗
脱物组分,确定目标组分的时间收集范围;富集目标组 intensity/cps 3.0×10 5
分,冷冻干燥后,称定质量,以最终获得CATM的物质的 2.0×10 5
量与灌流液中(S)-2-氧氯吡格雷的物质的量之比计算转
化率。 1.0×10 5
2.6 CATM的鉴定
0
2.6.1 质谱鉴定 取“2.5”项下制备的目标产物,用 15 20 25 30
50%甲醇溶解稀释成500 ng/mL,在正离子模式下,针泵 t/min
A.色谱图
进样,流速为 7 μL/min;在母离子全扫描模式下的扫描
范围为 350~360 Da,扫描速度为 10 Da/s,扫描时间为 5 8.0×10 6
min,分别确定目标产物母离子质荷比,并得最佳去簇电
压值;在子离子扫描模式下的初始碰撞能为 10 eV,以 6.0×10 6
5~10 eV为步长调节,扫描速度为200 Da/s,扫描时间为
5 min,获得子离子信息。 intensity/cps 4.0×10 6
2.6.2 NMR谱鉴定 以氘代二甲基亚砜[核磁共振氢谱
2.0×10 6
1
(H-NMR)为 2.50 ppm]为溶剂分别测定目标产物的
H-NMR谱和重水交换谱。将各氢信号分类编号汇总并 0
1
依据NMR命名法(化学位移、氢分布、峰分裂数)对分子 0 50 100 150 200 250 300 350 400
m/z
的单个质子氢信号进行共振分配,通过重水交换谱验证 B.质谱图
活泼氢信号。 图2 分离纯化后目标组分的色谱图和质谱图
·1726 · China Pharmacy 2022 Vol. 33 No. 14 中国药房 2022年第33卷第14期