
Page 79 - 202014
P. 79
Y=27 811X-7 826.8(r=0.999 9),乙酰紫草素的回归 3),表明该方法能够满足试验要求,耐用性良好。
方程为 Y=31 222X-95 144(r=0.999 8),β,β′-二甲基 2.5.11 样品含量测定 取 34 批药材样品粉末适量,按
丙烯酰阿卡宁的回归方程为 Y=34 776X-94 892(r= “2.5.2”项下方法制备供试品溶液,再按“2.5.1”项下色谱
0.999 9);上述3种成分检测质量浓度的线性范围分别为 条件进样测定,记录峰面积并按标准曲线法计算样品含
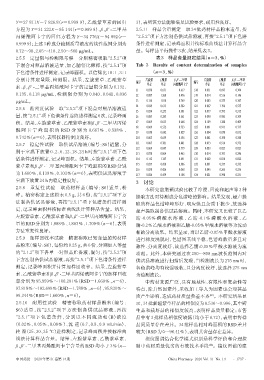
0.72~90、2.05~410、2.50~500 µg/mL。 量。每样品平行操作3次,结果见表3。
2.5.5 定量限与检测限考察 分别精密吸取“2.5.2”项 表3 样品含量测定结果(n=3,%%)
下混合对照品溶液适量,加乙腈倍比稀释,按“2.5.1”项 Tab 3 Results of content determination of samples
下色谱条件进样测定,记录峰面积。以信噪比10∶1、3∶1 (n=3,%%)
分别计算定量限、检测限。结果,左旋紫草、乙酰紫草 左旋紫 乙酰紫 β,β′-二甲基 左旋紫 乙酰紫 β,β′-二甲基
编号 编号
素、β,β′-二甲基丙烯酰阿卡宁的定量限分别为 0.132、 草素 草素 丙烯酰阿卡宁 草素 草素 丙烯酰阿卡宁
S1 0.038 0.651 0.617 S18 0.031 0.695 0.304
0.135、0.118 µg/mL,检测限分别为 0.040、0.041、0.036 S2 0.087 1.260 0.476 S19 0.014 0.516 0.146
µg/mL。 S3 0.134 1.010 0.769 S20 0.043 0.555 0.347
S4 0.029 0.613 0.582 S21 0.067 1.196 0.535
2.5.6 精密度试验 取“2.5.2”项下混合对照品溶液适
S5 0.054 0.255 0.301 S22 0.077 0.795 0.315
量,按“2.5.1”项下色谱条件连续进样测定6次,记录峰面 S6 0.083 0.283 0.561 S23 0.090 0.985 0.309
积。结果,左旋紫草素、乙酰紫草素和β,β′-二甲基丙烯 S7 0.069 0.635 0.668 S24 0.109 1.388 0.409
S8 0.069 0.552 0.733 S25 0.081 0.913 0.717
酰 阿 卡 宁 峰 面 积 的 RSD 分 别 为 0.667% 、0.538% 、
S9 0.058 0.682 0.827 S26 0.004 0.050 0.032
0.742%(n=6),表明仪器精密度良好。 S10 0.063 0.659 0.536 S27 0.041 0.190 0.340
2.5.7 稳定性试验 取供试品溶液(编号:S6)适量,分 S11 0.067 0.781 0.881 S28 0.033 0.514 0.372
S12 0.069 0.847 0.779 S29 0.003 0.025 0.023
别于室温下放置 0、2、6、12、18、24 h 时按“2.5.1”项下色 S13 0.070 0.864 0.465 S30 0.014 0.393 0.246
谱条件进样测定,记录峰面积。结果,左旋紫草素、乙酰 S14 0.112 1.347 0.691 S31 0.002 0.030 0.022
紫草素和β,β′-二甲基丙烯酰阿卡宁峰面积的RSD分别 S15 0.057 0.420 0.306 S32 0.031 0.357 0.332
S16 0.030 0.426 0.068 S33 0.045 0.265 0.336
为 1.660%、0.139%、0.103%(n=6),表明供试品溶液于 S17 0.024 0.459 0.108 S34 0.021 0.398 0.231
室温下放置24 h内稳定性良好。 3 讨论
2.5.8 重复性试验 取药材样品(编号:S6)适量,粉 本研究前期预试验比较了冷浸、回流和超声等 3 种
碎。精密称定上述粉末 0.5 g,共 6 份,按“2.5.2”项下方
提取方法对待测成分色谱峰的影响。结果发现,超声提
法制备供试品溶液,再按“2.5.1”项下色谱条件进样测 取的样品色谱峰峰形好、响应强且杂质干扰少,故选择
定,记录峰面积并按标准曲线法计算样品含量。结果, 超声提取制备供试品溶液。同时,本研究又比较了以乙
左旋紫草素、乙酰紫草素和β,β′-二甲基丙烯酰阿卡宁含 腈 -0.05% 磷 酸 水 溶 液 、乙 腈 -0.1% 磷 酸 水 溶 液 、乙
量的RSD分别为1.888%、1.863%、1.709%(n=6),表明 腈-0.2%乙酸水溶液和乙腈-0.05%甲酸水溶液等为流动
方法重复性良好。 相的分离效果。结果显示,当以乙腈-0.05%甲酸水溶液
2.5.9 加样回收率试验 精密称取已知含量的药材样 进行梯度洗脱时,色谱图基线平稳,色谱峰数量多且对
品粉末(编号:S6),每份约0.25 g,共6份,分别加入等量 称性、分离度较好,故选择乙腈-0.05%甲酸水溶液为流
的“2.1.2”项下各单一对照品贮备液,混匀,按“2.5.2”项 动相。此外,本研究通过在 200~800 nm 波长范围内对
下方法制备供试品溶液,再按“2.5.1”项下色谱条件进样 供试品溶液进行扫描后发现,当检测波长为275 nm时,
测定,记录峰面积并计算加样回收率。结果,左旋紫草 各色谱峰均有较强吸收,且分离度较好,故选择 275 nm
素、乙酰紫草素和β,β′-二甲基丙烯酰阿卡宁的加样回收 为检测波长。
率分别为 95.959%~100.201%(RSD=1.669%,n=6)、 中药材来源广泛,具有地域性、有限性和复杂性等
97.818%~102.698%(RSD=1.788%,n=6)、95.831%~ 特点,除自然因素外,采收加工等人为因素也会对其品
99.344%(RSD=1.600%,n=6)。 质产生影响,造成药材质量参差不齐 。本研究结果显
[23]
2.5.10 耐用性试验 精密称取药材样品粉末(编号: 示,34批新疆紫草样品的相似度为0.536~0.996,其中野
S6)适量,按“2.5.2”项下方法制备供试品溶液,再按 生品和栽培品的相似度较高,表明样品质量稳定;市售
“2.5.1”项下色谱条件,分别以不同流动相(B)浓度 品中有 3 批样品相似度较低(均小于 0.72),表明市售样
(0.02%、0.05%、0.08%)、流 速(0.7、0.8、0.9 mL/min)、 品间质量存在差异。34批样品相对峰面积的RSD差异
柱 温(25、30、35 ℃)进样测定,记录峰面积并按标准曲 较大(RSD为0~96.41%),表明其含量存在差异。
线法计算样品含量。结果,左旋紫草素、乙酰紫草素、 指纹图谱结合化学模式识别是科学评价和合理控
[22]
β,β′-二甲基丙烯酰阿卡宁含量的 RSD 均小于 3%(n= 制中药材质量优劣的有效技术手段 。指纹图谱的建
中国药房 2020年第31卷第14期 China Pharmacy 2020 Vol. 31 No. 14 ·1737 ·