
Page 67 - 《中国药房》2026年8期
P. 67
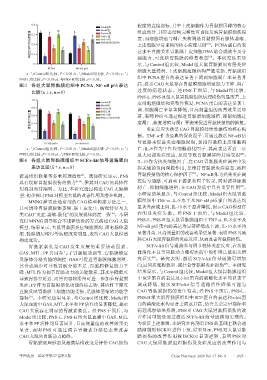
0.4 程度的直接指标,其中上皮细胞作为胃黏膜屏障的核心
Control组
bc a d Model组 组成部分,其形态结构完整性可直接反映胃黏膜损伤程
阳性区域平均光密度值 0.2 a b d b b bc PNS-H组 度;而细胞增殖与凋亡失衡则是胃黏膜固有腺体萎缩、
b
Positive组
0.3
PNS-L组
PNS-H+ISCK03组
[18]
上皮细胞异常重构的核心病理基础 。PCNA蛋白的表
达水平直接关系胃黏膜上皮细胞DNA的合成效率及分
0.1
[19]
示,与 Control 组比较,Model 组大鼠胃黏膜固有层炎症
0 裂能力,可反映胃黏膜的修复状态 。本研究结果显
PCNA NF-κB p65 细胞大量浸润,上皮细胞超微结构严重受损,胃黏膜组
a:与Control组比较,P<0.05;b:与Model组比较,P<0.05;c:与
PNS-L组比较,P<0.05;d:与PNS-H组比较,P<0.05。 织中 PCNA 蛋白的表达显著下调而细胞凋亡率显著升
图3 各组大鼠胃黏膜组织中 PCNA、NF-κB p65 表达 高,提示CAG大鼠存在胃黏膜细胞增殖能力下降、凋亡
比较(x±s,n=8) 过度的病理状态。经 PNS 干预后,与 Model 组比较,
PNS-L、PNS-H组大鼠胃黏膜组织病理损伤明显改善,上
1.0
Control组 皮细胞超微结构完整性恢复,PCNA蛋白的表达显著上
Model组
0.8 b bc Positive组 调、细胞凋亡率显著降低,且高剂量组的改善效果更显
蛋白相对表达量 0.6 b d b b bc d PNS-H组 著,表明 PNS 可通过促进胃黏膜细胞增殖、抑制细胞过
PNS-L组
PNS-H+ISCK03组
度凋亡、恢复增殖与凋亡平衡来促进胃黏膜损伤的修复。
0.4
0.2 a a 炎症反应失衡是CAG胃黏膜持续性损伤的核心机
制。TNF-α作为经典的促炎因子,可通过激活NF-κB信
0
SCF/β-actin p-c-kit/c-kit 号通路来促进炎症细胞浸润,加剧胃黏膜上皮细胞凋
a:与Control组比较,P<0.05;b:与Model组比较,P<0.05;c:与 亡;IL-8 作为中性粒细胞趋化因子,其高表达可进一步
PNS-L组比较,P<0.05;d:与PNS-H组比较,P<0.05。 放大局部炎症反应,从而导致胃黏膜屏障持续受损 ;
[20]
图4 各组大鼠胃黏膜组织中 SCF/c-kit 信号通路蛋白 IL-10 作为抗炎细胞因子,在 CAG 胃黏膜炎症调控中发
表达比较(x±s,n=8) 挥关键的负向调控作用,是维持胃黏膜炎症稳态、减轻
[21]
[3]
促进组织修复等多重药理活性 。既往研究显示,PNS 胃黏膜损伤的核心保护因子 。NF-κB作为经典炎症调
具有缓解胃黏膜损伤的潜力 [5,15] ,但其对 CAG 的具体作 控信号通路,可启动下游促炎因子转录,同时诱导细胞
[22]
用机制尚待阐明。为此,本研究通过构建 CAG 大鼠模 凋亡、抑制细胞增殖,在 CAG 发病中具有重要作用 。
型,初步探讨PNS对模型大鼠的改善作用及潜在机制。 本研究结果显示,与 Control 组比较,Model 组大鼠胃黏
MNNG 灌胃法是常用的 CAG 模型构建方法之一, 膜组织中 TNF-α、IL-8 水平及 NF-κB p65 蛋白的表达均
其可诱导胃黏膜腺体萎缩、肠上皮化生,病理特征与人 显著升高或上调,IL-10水平显著降低,提示CAG模型存
[16]
类CAG“炎症-萎缩-肠化”的发展规律高度一致 。本研 在明显炎症失衡。经 PNS 干预后,与 Model 组比较,
究以MNNG诱导联合不规律饮食的方式构建CAG大鼠 PNS-L、PNS-H组大鼠胃黏膜组织中TNF-α、IL-8水平及
模型,结果显示,大鼠胃黏膜炎症细胞浸润、固有腺体萎 NF-κB p65蛋白的表达均显著降低或下调,IL-10水平均
缩、黏膜肌层增厚等病理改变明显,表明CAG大鼠模型 显著升高,且高剂量组的改善效果更显著,表明PNS可减
构建成功。 轻CAG大鼠胃黏膜的炎症反应,从而改善胃黏膜损伤。
胃激素紊乱是 CAG 发生发展的重要始动因素, SCF/c-kit信号通路参与调节机体炎症反应,在功能
GAS、MTL、PP 共同参与了胃肠蠕动调节、胃黏膜修复 性消化不良等胃肠动力障碍疾病中的作用已被相关研
[23]
及腺体分泌功能的调控:GAS 可促进胃黏膜细胞增殖, 究证实 。研究表明,激活 SCF/c-kit 信号通路可增加
[24]
其合成减少可导致胃酸分泌不足、胃黏膜修复能力下 Cajal间质细胞数量、减轻食管黏膜炎症损伤 。本研究
降;MTL 作为调节胃肠动力的关键激素,其水平降低可 结果显示,与 Control 组比较,Model 组大鼠胃黏膜组织
导致胃排空延迟,而胃内容物滞留可进一步加重胃黏膜 中 SCF 蛋白的表达及 c-kit 蛋白的磷酸化水平均显著下
炎症;PP 作为胃黏膜消化功能的标志物,其活性下降可 调或降低,提示 SCF/c-kit 信号通路活性降低可能与
直接反映胃黏膜主细胞功能受损,是腺体萎缩的功能学 CAG 胃黏膜损伤的发生有关;经 PNS 干预后,PNS-L、
[17]
指标 。本研究结果显示,与 Control 组比较,Model 组 PNS-H 组大鼠胃黏膜组织中 SCF 蛋白的表达和 c-kit 蛋
大鼠血清中GAS、MTL水平和PP活性均显著降低,提示 白的磷酸化水均显著上调或升高,结合其余定量指标和
CAG 大鼠存在明显的胃激素紊乱。经 PNS 干预后,与 病理观察结果推测,PNS 对 CAG 大鼠胃黏膜损伤的改
Model 组比较,PNS-L、PNS-H 组大鼠血清中 GAS、MTL 善作用可能是通过激活 SCF/c-kit 信号通路而实现的。
水平和 PP 活性均显著回升,且高剂量组的改善效果更 为验证上述推测,本研究在高剂量PNS的基础上联合通
显著,表明 PNS 可通过调节胃激素分泌稳态来改善 路抑制剂ISCK03进行干预,结果显示,PNS对大鼠胃黏
CAG大鼠的胃肠动力障碍。 膜损伤的改善作用被 ISCK03 显著逆转,表明 PNS 对
胃黏膜病理形态及超微结构改变是评价CAG损伤 CAG 大鼠胃黏膜组织损伤及炎症反应的改善作用与
中国药房 2026年第37卷第8期 China Pharmacy 2026 Vol. 37 No. 8 · 1025 ·